






Published online Nov 27, 2021. doi: 10.4254/wjh.v13.i11.1552
Peer-review started: February 27, 2021
First decision: May 2, 2021
Revised: May 7, 2021
Accepted: August 20, 2021
Article in press: August 20, 2021
Published online: November 27, 2021
Processing time: 269 Days and 16 Hours
Chelation is the mainstay of therapy in certain pediatric liver diseases. Copper and iron related disorders require chelation. Wilson’s disease (WD), one of the common causes of cirrhosis in children is treated primarily with copper chelating agents like D-penicillamine and trientine. D-Penicillamine though widely used due its high efficacy in hepatic WD is fraught with frequent adverse effects resulting discontinuation. Trientine, an alternative drug has comparable efficacy in hepatic WD but has lower frequency of adverse effects. The role of ammonium tetra-thiomolybdate is presently experimental in hepatic WD. Indian childhood cirrhosis is related to excessive copper ingestion, rarely seen in present era. D-Penicillamine is effective in the early part of this disease with reversal of clinical status. Iron chelators are commonly used in secondary hemochromatosis of liver in hemolytic anemias. There are strict chelation protocols during bone marrow transplant. The role of iron chelation in neonatal hemochromatosis is presently not in vogue due to its poor efficacy and availability of other modalities of therapy. Hereditary hemochromatosis is rare in children and the use of iron chelators in this condition is limited.
Core Tip: Chelation forms the most important part of management of certain liver diseases in children. In Wilson's disease and secondary hemochromatosis related to transfusion, chelation is well established treatment modality with proven efficacy. In other diseases like copper associated childhood cirrhosis and neonatal hemochromatosis the role of chelation is doubtful. In hereditary hemochromatosis, chelation is recommended as alternative therapy. The selection of chelating agents for treatment depends on the efficacy, feasibility and risk of adverse effects known from literature. The review discusses the concepts of chelation and reviews the literature to assess the role of chelation in treatment of various pediatric liver diseases.
- Citation: Seetharaman J, Sarma MS. Chelation therapy in liver diseases of childhood: Current status and response. World J Hepatol 2021; 13(11): 1552-1567
- URL: https://www.wjgnet.com/1948-5182/full/v13/i11/1552.htm
- DOI: https://dx.doi.org/10.4254/wjh.v13.i11.1552
Chelation is a process in which a synthetic compound is administered to remove an excess mineral or heavy metal from the body. There are various liver diseases that are caused by excess deposition of various heavy metals such as copper, iron and arsenic. Some of these are genetic-metabolic, others are due to environmental exposure. In the landmarks of chelation therapy in hepatology, Walshe documented cupriuresis after administering dimethyl cysteine (penicillamine) in Wilson’s disease (WD) in 1956[1]. Chelation was thereafter used in non-Wilsonian liver diseases. In the subsequent years newer chelators such as trientine and ammonium tetra thiomolybdate were identified for WD. From the 1970s, transfusion-related liver siderosis of hemolytic anemias was revolutionized by the use of deferoxamine[2]. The use of iron chelators was attempted in gestational alloimmune liver disease and hereditary hemochromatosis. This review explores the rationale and outcome of chelation therapy in various pediatric liver diseases.
Metal ion (M) complexes with cheating agent (L) through an equilibrium reaction to form metal-ligand complex (ML) or chelate. The concentration of the chelate in the solution is directly proportional to the concentration of metal ion [M] and the ligand [L].
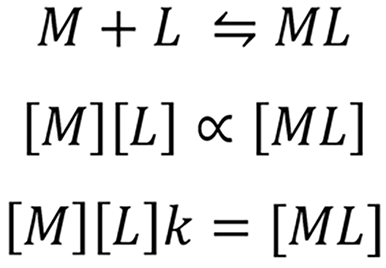
Where k is the effective stability constant. Value k denotes the affinity of the chelating agent. High k values suggest high affinity of the chelating agent. The value of k depends on the nature of the chelating agent, temperature, pH of the solution[3]. The in-vivo milieu is not similar to the in-vitro chemical reaction. The presence of weak acids in the body fluids like glutamate, sulfate, citrate, amino acids, albumin, macroglobulin etc. affect the chelation. These are called biological ligands. Chelating agent binds to the biological ligands and the effective concentration in the body fluid is lowered. Hence the equation becomes.
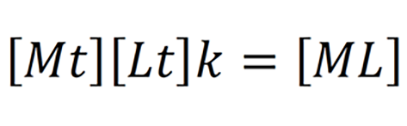
Where Mt, Lt is the total concentration of the metal ion and chelating agent respectively which is very difficult to assess in the clinical setting[4].
Effective chelation occurs when concentration of M and/or L is high, when affinity of the chelator (k) is high or when the concentration of the chelate [ML] is low. The metal ion concentration [M] in the body depends on the severity of the disease. For example, in a WD presenting as acute liver failure, serum copper (Cu) levels are usually very high. The concentration of chelating agent [L] is increased by increasing the dosing and/or frequency as tolerated by the patient. For the chelation to progress, urinary excretion of chelate [ML] is very important as it effectively reduces the concentration[3]. Ideal chelating agents must have good oral absorption, acceptable bioavailability, high affinity to metal ions, low toxicity at appropriate plasma concentration, undergo rapid elimination or detoxification after combining with metal ions and more importantly should be available in affordable price[5].
WD is an autosomal recessive disorder caused by mutation of ATP7B gene that encodes for a protein P-type ATPase which transports copper into trans Golgi network and for biliary excretion of copper. In lysosomes, copper is incorporated into ceruloplasmin. In WD, due to defect in ATPase transport protein, ceruloplasmin formation is defective and biliary excretion of copper is impaired[6,7]. This causes excess accumulation of intracellular copper subsequently increasing the levels in blood causing accumulation in extra-hepatic organs (Figure 1).

D-Penicillamine (3, 3-dimethylcysteine) is the most commonly used medication for WD worldwide. The L-isomer of this drug is not advised for treatment due to its neurotoxicity. The chelation property of DPA is due to the presence of thiol (-SH), which is responsible for its high affinity towards divalent metal ions such as copper. The mechanism of action of D-Penicillamine (DPA) is by inducing cuprieuresis, inducing hepatic metallothionine synthesis, reducing fibrosis (by preventing collagen formation). DPA also has an anti-inflammatory property[8]. It is rapidly absorbed in proximal intestine but only 40%-70% are absorbed[9]. The peak plasma concentration occurs after 1-3 h after ingestion. It circulates in the plasma predominantly by binding to albumin (80%), while the rest of the compound is present as free or disulphide forms. DPA is metabolized in the liver by conjugation with sulfide or by methylation (phase II reaction) and excreted in urine with almost 80% being eliminated within 10 h of ingestion. After discontinuation of therapy, the drug is eliminated in about 3-6 d[10]. Food, antacids, iron and zinc preparations reduce the bioavailability by almost 50%. Plasma concentration reduces significantly when the drug is taken with food[11]. It is recommended to give the drug either 1- hour before or 2- h after food. The drug is given in the dose of 20 mg/kg per day (up to 1500 mg) rounded to nearest 250 mg in 2-4 divided doses and can be maintained at 1000 mg/d once the disease is in remission[12]. As DPA causes pyridoxine deficiency, pyridoxine should be supplemented at 25-50 mg/d. In case of neurological WD, to prevent paradoxical neurological worsening, the drug is started at low dose (125-250 mg) and slowly increased (125-250 mg every week) to reach the desired dose by 4-6 wk[13].
Trientine (triethylenetetramine) is an alternative chelating agent in WD. It is a derivative of spermine and putrescine and binds to copper in the ratio 1:1 to form a stable complex, which is eliminated in the urine. Trientine dihydrochloride is the oral ingestible form requiring storage at 2-8 degree Celsius to maintain stability. 10% of the trientine is absorbed in the proximal small intestine and achieves its peak concentration 1.5-4 h after ingestion. Trientine is extensively metabolized in tissues by acetylation but the enzyme responsible for it is not identified. 1% of ingested trientine and 8% trientine metabolite acetyltrien, appears in the urine. Plasma concentration of the trientine significantly reduces when given with food due to its affinity to dietary copper in the lumen thereby compromising the removal of tissue copper and the other reason could be due to the physiological polyamines secreted during food intake inhibits effective trientine absorption[14]. Trientine is not to be given with iron as it forms toxic complexes. The dose recommended is 20 mg/kg per day with the maximum of 1500 mg/d rounded to nearest 250 mg (300 mg capsules in North America) and maintenance dose of 1000 mg/d. Similar to DPA, trientine also should be ingested 1 h before or 2 h after food intake[12,15]. The decoppering efficacy of any chelating agent is evident from the effective stability constant (k) which denotes copper affinity. The comparison of k-value of DPA (2.38 × 10-16) and trientine (1.74 × 10-16) suggests the decoppering efficacy of DPA is much higher than trientine[16].
Improvement in symptoms and biochemical parameters in WD takes around 2-6 mo in hepatic forms whereas in isolated neurological forms it may take up to 12-24 mo[12]. DPA in WD children shows an efficacy of almost 70%-90%[17-20]. The response depends on whether it is hepatic or neurological form and severity of the disease at presentation. Long term of follow up of WD (median duration- 15.1 years) studied by Bruha et al[19] showed the response to DPA to hepatic forms is 82% compared to 69% for neurological forms. One of the largest series of WD patients (n = 327) from Euro Wilson consortium, showed hepatic forms had 91% response compared to only 68% in neurological forms after a median follow up duration of 13.3 years[20]. In most series, trientine is used as a second line either due to poor response or due to toxicity to DPA. Hence, there are no head-to-head randomized trials comparing the efficacy of DPA and trientine. Overall efficacy of trientine is reported to be 80%-92%[21,22]. Retrospective analysis of efficacy of the two drugs by Hölscher et al[23] showed response in hepatic forms with DPA was 92% compared to 84% response with trientine after a median follow up duration of 13.3 years. In neurological forms, DPA fares significantly better (68%) than trientine (48%, P = 0.008)[23]. In Euro Wilson consortium, the response of both the DPA and trientine were comparable when used as a first line in both hepatic (90.7% vs 92.6%, P = 0.98) and neurological forms (67.5% vs55%, P = 0.76). However when used as a second line therapy, trientine vs DPA showed similar response in hepatic form (75% vs 68.9%, P = 0.76) but better response in neurological form (51% vs 23.1%, P = 0.01)[20].
Adverse effects of DPA are always a major concern with up to 30% of the patients develop one or more adverse effects (Table 1)[20,24,25]. Adverse effect can be early onset (less than 3 wk of therapy) or late (more than 3 wk to up to 2-3 years of initiation of therapy). Early adverse effects like fever, rash, arthralgia, lymphadenopathy, pancytopenia are predominantly immune mediated[26]. Nephropathy, the most common late adverse effect of DPA is seen in 5%-30%. Presentations include proteinuria, glomerulonephritis, nephrotic syndrome less commonly as Good Pasture’s syndrome[27-29]. More than 90% of the nephropathy occurs within 12 mo of therapy. High doses of DPA, decompensated liver disease, intrinsic renal diseases or presence of HLA-B8/DR3 are probable risk factors of nephropathy[30]. Eighty percent are membranous glomerulonephritis on renal biopsy. In a study by Hall et al[27] of 33 patients with DPA nephropathy, one-third each showed resolution at 6, 12 and 18 mo respectively, after drug discontinuation. There are no clear recommendations as to whether the drug can be rechallenged after resolution of nephropathy. However, in such situations, it is prudent to continue the patient on an alternative drug such as trientine or zinc. DPA related myelotoxicity occur in up to 7% patients undergoing chelation with DPA[31-33]. Two types of myelotoxicity are known to occur, idiosyncratic (usually with in 1 year of therapy) or dose dependent (more than after 1 year therapy)[34]. Though, there are no definite guidelines for monitoring and treatment of myelotoxicity, European society of Pediatric Gastroenterology Hepatology and Nutrition (ESPGHAN) suggests weekly blood counts initially, 1-3 mo till remission and 3-6 monthly thereafter[35]. If two or more values of total leukocyte count less than 3.5 × 103 per cubic mm, drug is to be discontinued. Bone marrow examination and reticulocyte counts differentiates this condition if concomitant hypersplenism is present[36,37]. Blood products, colony stimulating factor and anti-thymocyte globulin may improve the counts. Usual time of spontaneous recovery is 4-12 wk. Rarely hematopoietic stem cell transplantation may be required in refractory and prolonged cases. Once bone marrow toxicity has ensued, the drug should not be re-challenged. Adverse effects of DPA related to skin may be due to either acute hypersensitivity reaction presenting as morbilliform rash, urticaria, degenerative dermatoses (cutis laxa or elastosis perforans serpingosa) or an autoimmune phenomenon (pemphigus, scleroderma or lichen planus[38]. Rare musclar adverse effects of DPA include myasthenia (1%-2%) and ptosis. Anti- nicotinic acetyl choline receptor or Anti- MuSK (Anti- Muscle Specific tyrosine Kinase) is present in up to 70%[39]. Systemic lupus erythematosus can occur within 6-12 mo after the onset of DPA therapy presenting as pleurisy, arthritis, rash with or without presence of anti-nuclear antibody[40]. Deutscher et al[41] noted 3 out of 50 WD children with elevated transaminases within 6 wk of DPA therapy who resolved subsequently following discontinuation. Trientine also present with similar adverse effects as DPA like nausea, vomiting, arthralgia, myalgia, leukopenia, elevation in anti-nuclear antibody (ANA), nephropathy but adverse effects requiring discontinuation of trientine is significantly lower compared to DPA[20].
| Name of the drug | Side effects |
| D-Penicillamine | Early (1-3 wk): Fever, rash, arthralgia, cytopenia, proteinuriaLate: (1) Skin: degenerative dermatoses elastosis perforans serpingosa, cutis laxa, pseudoxanthoma elasticum, bullous dermatoses, psoriasiform dermatoses, lichen planus, seborrheic dermatitis alopecia, aphthous ulcerations, hair loss; (2) Connective tissue disorders: Lupus like syndrome, arthralgia, Rheumatoid arthritis, polymyositis; (3) Renal: proteinuria, hematuria, glomerulonephritis, nephrotic syndrome, renal vasculitis, Goodpasture’s syndrome; (4) Nervous system: paradoxical neurological worsening, neuropathies, myasthenia, hearing abnormalities, serous retinitis; (5) Gastrointestinal: Nausea, vomiting, diarrhea, elevated transaminases, cholestasis, hepatic siderosis; (6) Respiratory: pneumonitis, pulmonary fibrosis, pleural effusion; (7) Hematological: cytopenia, agranulocytosis, aplastic anemia, hemolytic anemia; and (8) Others: Immunoglobulin deficiency, breast enlargement, pyridoxine deficiency |
| Trientine | Paradoxical neurological worsening (10%-50%), sideroblastic anemia, bone marrow suppression, gastritis, skin rash, arthralgia, myalgia, hirsutism |
| Ammonium tetra thiomolybdate | Neurological dysfunction (rare), hepatotoxicity, bone marrow suppression |
In hepatic WD, paradoxical neurological worsening occurs commonly within 6 mo of therapy, in patients with an underlying overt or occult neuropsychiatric feature. Paradoxical neurological worsening occurs even when dosing and compliance is good[42]. It occurs due to the sudden release of Cu from the liver following chelation therapy causing oxidative brain injury. Overall incidence of paradoxical neurological worsening ranges from 7%-26%. Those with previous known neurological WD, the incidence of worsening is up to 75%[19,24,25]. Both DPA and TA have shown to cause neurological worsening. In series from Euro Wilson consortium, paradoxical neurological worsening occurred significantly more with TA compared to DPA[20]. Litwin et al[13] studied natural history of 143 WD (70 Neuro/Neurohepatic WD and 73 hepatic WD), of whom 23% neurological cohort and none of the hepatic cohort developed early neurological worsening on chelation. In this series, median time of onset of neurological worsening was 2.3 mo. Fifty-three percent were completely reversible and 13% were partially reversible on drug discontinuation with median time of reversibility of 9.2 mo[13]. Prior neurological involvement, lesions in brain stem or thalamus and concomitant anti-dopaminergic drugs had higher chances of neurological worsening. Treatment consists of drug discontinuation and addition of zinc for a transition period. Chelators can be restarted in lower doses with gradual increment once the symptoms improve[13].
Currently there is no fool-proof, gold standard yardstick to assess chelation adequacy. All have fallacies in assessment and hence multiple parameters are considered. Chelation adequacy can be assessed firstly by assessing compliance to drug intake. Compliance is assessed by having a pill count, self-reporting by patients themselves or by checking empty blister packs during follow up outpatient visits[43]. There are various scales being developed assessing medication adherence (MAQ: Medication adherence questionnaire, MARS: Medication adherence Rating scale) but none have been validated in children[44]. More objective way of assessing compliance is by measuring drug levels but it is not routinely available under clinical setting. Secondly, follow up of clinical parameters assess the adequacy of chelation like improvement in jaundice, ascites, encephalopathy which usually take 2-6 mo post therapy. Resolution of neurological symptoms may take longer than 2-3 years[12]. The resolution of Kayser-Fleischer ring on de-coppering therapy has considerable controversies to the same. Studies have heterogeneity in their assessment and reports. It appears to be independent on type of presentation (neurologic vs hepatic), stage of disease (pre-symptomatic vs symptomatic) and choice of chelator and compliance. Initial reports showed, Kayser-Fleischer (KF) ring disappearance in 81% of the patients (completely in 41% and incompletely in 59%), more in pre-symptomatic stage (60%) than those in symptomatic phase with ongoing therapy (2%) over 22 years of follow-up on DPA (90%) and zinc or trientine (10%). Conversely one-third of asymptomatic patients the rings did not reabsorb even after therapy of > 10 years. In this study, the fading of KF rings seemed to be independent of the stage of the disease and effectiveness of the decopperizing treatment[45]. In a study by Fenu et al[46] where 66% were hepatic and 31% were neuro-hepatic (90% on DPA ± zinc therapy), partial or total KF ring resolution was observed in 28%, deterioration in 6% and static in the rest of the cohort over 1-3 years of therapy. Other smaller cohorts report reduction of KF ring in neuropsychiatric manifestation or disappearance over 10 years on maintenance zinc and molybdate therapy in pediatric hepatic WD[47,48]. KF rings may reappear with non-compliance, and occasionally even with successful maintenance therapy[49].
Liver status can be appropriately assessed by Pediatric end-stage liver disease or Child-Turcotte-Pugh score. Biochemical parameters like serum albumin, total bilirubin and prothrombin time normalizes by 6 mo but liver enzymes might take longer[12]. In the author’s experience it takes 9-12 mo for complete normalization of Liver function tests in majority of the cases[50]. In patients who have additional neurological involvement, neurological response is monitored by indices such as Global assessment scale (GAS)[51]. Even with neurological WD with significant MRI changes, 50% show improvement with long term chelation[52].
Presently the most widely acceptable way to assess adequacy of chelation is by 24-h urine copper and non-ceruloplasmin copper. Twenty-four hours urine copper (UCu) increases immediately following chelation and takes around 12-18 mo to reach a stable level[53]. European Association for the Study of the Liver (EASL) and American Association for the Study of Liver diseases (AALSD) recommends targeting 24-h urine copper between 200-500 mg/d for adequate chelation[12,15]. Values > 500 mg/d suggest under chelation as lot of unchelated copper is remaining in the body. Values < 200 mg/d may be either due to over chelation or poor compliance (Table 2). This can be differentiated by non-ceruloplasmin copper (NCC) levels calculated by the formula (serum copper (mg/L) - 0.3 x serum ceruloplasmin(mg/L)[54]. NCC has a few fallacies. Firstly, almost 20% of NCC are negative values, seen mostly when immunoassay method was used to measure ceruloplasmin as it measures both holoceruloplasmin and apoceruloplasmin. NCC calculation becomes inappropriate when inactive apoceruloplasmin is included. Secondly, there are variabilities in reference ranges in ceruloplasmin values between various laboratories across the world creating disparities in NCC cut-offs[55]. According to EASL guidelines, NCC > 15 mg/dL suggest poor compliance and < 5 mg/dL suggest over chelation. Additionally, 24-h urine copper after 48-h cessation of therapy has been recommended by EASL. Values > 100 mg/d is suggestive of under chelation or poor compliance while values < 100 mg/d suggest adequate treatment[15].
| Early stages of treatment (< 1 yr) | UCu > 500 μg/dNCC > 25 μg/dL |
| Good control (treatment > 1 yr) | UCu 200-500 μg/dNCC < 15 μg/dL |
| Poor compliance/uncontrolled disease | UCu > 500 μg/dNCC > 15 μg/dL |
| Inadequate dose | UCu < 200 μg/dNCC > 15 μg/dL |
| Over-treatment | UCu < 200 μg/dNCC < 5 μg/dL |
A novel and upcoming modality to assess chelation is the use of exchangeable copper. Exchangeable copper is the fraction of copper bound to albumin, peptide and amino acids which are easily chelated by chelating agents. It denotes a direct estimation of non-ceruloplasmin copper (NCC)[56]. On WD with chelation for long time, exchangeable copper values tend to reduce comparable to non-Wilson children. In a pilot study by the authors, the role of exchangeable copper was assessed in a cohort of 96 children with hepatic WD. Exchangeable copper was significantly higher in newly diagnosed WD compared to WD on chelation for more than 1 year (3 ± 7 μmol/L vs 0.9 ± 0.6 μmol/L, P = 0.03). Exchangeable copper values were lower in stable liver disease compared to unstable liver disease (0.86 ± 0.5mmol/L vs 1.3 ± 0.6 mmol/L, P = 0.01). Exchangeable copper values showed excellent correlation with non-ceruloplasmin copper (r = 0.92, P < 0.001). Predictive model incorporating exchangeable copper into standard monitoring tools improved the yield of disease control assessment by 21%[57].
Strictly zinc is not considered as a systemic chelator. Oral zinc (Zn) induces metallothionine in enterocyte. Metallothionine is an endogenous chelator that has high affinity to copper. Hence induced metallothionine combines with luminal Cu, preventing its entry into circulation. This Cu is removed through feces when enterocyte is shed. Zn also induces hepatic metallothionine[58]. Hence, Zn is used in pre-symptomatic WD, stable well chelated WD on maintenance therapy, severe neurological WD. It is also used as a last resort in those with DPA or trientine intolerance. In severe hepatic disease, many centers consider giving a trial of dual chelation DPA and zinc for rapid chelation and quick stabilization. In a study conducted by the authors, 65 children with > 9 mo chelation were followed up for long term outcome. Majority had advanced disease at presentation. 83% of children were treated with DPA mon
Ammonium tetra thiomolybdate is a strong decoppering agent used in limited trials. It prevents intestinal absorption of copper if given with meals but also reduces serum copper when given in between meals. Ammonium tetra thiomolybdate (ATM) is predominantly advised for neurological forms due to it low risk of neurological worsening[65]. In the comparative study of ATM with trientine in neurological WD, paradoxical neurological worsening is significantly lower with ATM (4%) compared to trientine (26.1%, P = 0.01)[66]. At larger doses, ATM can form toxic insoluble complex that gets deposited in liver causing hepatoxicity[67]. Hence the role of ATM in hepatic WD is precarious. Up to 10% of patients receiving ATM might develop bone marrow toxicity also[68]. Bis-choline tetra thiomolybdate (WTX101) is an investigational derivative of ATM being studied recently in neurological WD with better stability and lower toxicity[69]. Twenty-four weeks treatment of the drug caused improvement in 71% of neurological WD. Seven percent developed leukopenia and almost 39% developed elevated liver enzymes post therapy[69]. Robust experience in exclusive hepatic WD is not yet available.
Indian childhood cirrhosis is commonly seen in children between 6 mo and 5 years of age in Indian subcontinent with its peak incidence seen during 1970-1990[70]. Presently this entity seems to be waning in the Indian subcontinent. Predominant etiology advocated was excessive copper ingestion with use of copper utensils[71]. There was also a possibility of genetic predisposition affecting copper metabolism[70]. Clinical features consist of nonspecific symptoms to start with like fever, lethargy, easy fatiguability, palpable liver with leafy edges in stage I, splenomegaly and ascites in stage II and jaundice, coagulopathy and encephalopathy in stage III. Histopathological examination of liver shows diffuse hepatocyte necrosis, presence of Mallory bodies and granular orcein staining. Treatment monitoring is by liver function tests (LFT), serum copper and in many studies, by repeat hepatic copper and liver histology, while on treatment. Mortality is almost 60% in stage II but reaching almost 90% in stage III[72]. In the study by Bavdekar et al[73] 65 children with Indian childhood cirrhosis (ICC) on treatment with DPA were followed up for the mean duration of 3.5 years, showed response in 60% of the children in pre-icteric phase compared to only 6% response (P < 0.01) in icteric phase (Table 3). Another study in ICC children who received DPA or DPA with steroids showed 50% survival as compared to10% in placebo group (P = 0.002)[74].In a pediatric study, DPA therapy has showed better response compared to DPA with intravenous immunoglobulin (P = 0.018)[75]. Chelation may improve symptoms if given early as prognosis is poor in advanced disease despite treatment[75].
| Ref. | Disease | Drug | Follow up duration | Response | Adverse effects |
| Dhawan et al[60] | WD | DPA (n = 32) | Median:11.78 (1.45-34.2) yr | 20/32 (62.5%) | Minor- 6.3%; Major- 21.9% |
| Wang et al[106] | WD | DPA/TA (n = 9) | Mean: 5.1 4.1 yr | All responded | Not mentioned |
| Das et al[50] | WD | DPA (n = 65), TA(n = 4) | Median: 3.6 (0.8-12) yr | DPA (42/65) 64.6%, TA (3/4) 75% | DPA 10.8% |
| Arnon et al[107] | WD | TA (n = 10) | Treatment duration: 18 mo. Follow up:12-60 mo | All responded | 1/10 (10%) reported hepatotoxicity |
| Taylor et al[108] | WD | TA (n = 16) | 6.4 (0.78-18.6) yr | 14/16 (87.5%) | 1 had allergic reaction |
| Santos Silva et al[59] | WDAll decompensated liver disease | DPA (n = 1)TA (n = 4) | 18-60 mo | All responded one still had raised transaminase | 3/4 (75%) on DPA developed cytopenia |
| Bavdekar et al[73] | ICC | DPA (n = 68) | 3.5 (1-7) yr | 29/68 (42.6%) alive after follow up | 5 children had proteinuria |
| Tomar et al[75] | ICC | DPA (n = 60) | 12 mo duration | 13/17 (76.5%) of grade III survived | 11.8% drug rash, 5.9% fever |
| Tanner et al[74] | ICC (15 children treated with DPA in both trials together) | DPA (n = 15) | 6 yr | Trial I: 1/15 (6.7%) survived in 6 yr, Trial II: 5/10 (50%) survived in 6 yr | Not mentioned |
| Horselen et al[77] | Case report CACC (age 7 yr) | DPA | 19 mo | Hepatic copper normalized | none |
| Maggiore et al[78] | Case report CACC (age 10 yr) | DPA | 24 mo | No improvement | Not mentioned |
| Rodeck et al[109] | CACC (age 6 and 10 mo) | DPA | 18 mo, other child deteriorated immediately following DPA initiation | One child improved and other developed acute liver failure requiring liver transplantation | None |
| Flynn et al[83]2002 | NH | DFO (n = 5) with antioxidant | Follow up at 48 mo | 2/5 (40%) survived without transplantation | Not mentioned |
| Rodrigues et al[84] 2005 | NH | DFO with antioxidant (n = 9) | Follow up 3-9.8 yr | 1/9 (11.1%) survived without transplantation | Not mentioned |
| Sigurrdson et al[85] 1998 | NH | DFO with antioxidant (n = 8) | Not mentioned | None survived without transplantation | Not mentioned |
| Masera et al[110] 2013 | HJV hemochromatosis Case report (7/F) | DFX | 12 mo of treatment | Iron indices improved on 12 mo treatment | Not mentioned |
Non-Wilsonian copper related diseases termed by Baker et al[76] as copper associated childhood cirrhosis includes ICC from India and ICC-like illness from western countries. This ICC like illnesses is otherwise called idiopathic copper toxicosis. Type I copper associated childhood cirrhosis (CACC) resembles ICC, with an early onset of disease and related to increased copper intake. Type II CACC has onset later than 4 years of age and possibly has an autosomal recessive inheritance without an obvious increase in copper intake[77]. Although there are few case reports of ICC- like illnesses, meagre number of reports use chelation therapy probably due to its conflicting results. One child from Bangladeshi origin, presented with jaundice, anorexia, weight loss at 7 years, with normal serum ceruloplasmin, and elevated hepatic copper 2319 mg/g. Improvement in symptoms and decrease in liver copper (35 mg/g) was noted after 19 mo of DPA therapy (Table 3)[77]. In contrast, a 10 year old Italian child with ascites and hepatomegaly, normal ceruloplasmin levels and liver copper of 1970 mg/g did not show any improvement clinically and biochemically even after 2 years of DPA[78]. Largest cohort of endemic Tyrolean infantile cirrhosis studied by Muller et al[79] showed both genetics and copper contamination were responsible for the disease. However there is paucity of chelation therapy experience in this condition.
In Gestational alloimmune liver disease alloimmunization of fetal liver antigen occurs in maternal blood resulting in IgG fetal liver antibody causing complement activation in fetal liver and significant impairment in hepcidin production (Figure 2)[80]. This causes iron storage in various organs like liver, heart, gonads, pancreas etc. Gestational alloimmune liver disease (GALD) causes liver failure as a result of hemochromatosis in newborn period and has high mortality if not intervened earlier. The liver injury causes reduced production of hepcidin resulting in uncontrolled iron absorption through placenta. This excess iron might further aggravate liver injury and also result in extra-hepatic iron deposition[81,82]. There have been few studies of GALD being treated with iron chelators (intravenous deferoxamine) and antioxidants with no clear-cut benefit. In the series by Flynn et al[83] five infants with neonatal hemochromatosis received intravenous deferoxamine but only one survived without liver trans

Hemochromatosis is due to iron accumulation in various organs with secondary causes being commoner in children than hereditary hemochromatosis. Secondary causes of hemochromatosis are commonly related to repeated transfusions in hemolytic anemia especially thalassemia major. In normal individuals, increased plasma iron induces the genes like HFE, TFR2 and HJV. This causes release in hepcidin, binding with ferroportin in enterocytes and macrophages, reducing iron absorption. Hereditary hemochromatosis (HH), most commonly due to mutation in HFE, cause impaired production of hepcidin making checkpoint for iron absorption defective[86]. Animal studies showed excessive fat intake causes impaired hepcidin production and increased transferrin receptor 1 and divalent metal transporter 1 Levels by altering mRNA expression. Hence, increased iron absorption and iron related liver injury may be responsible for development of non-alcoholic steatohepatitis[87]. Hereditary hemochromatosis (HH) is extremely rare in children. Excess iron in the serum causes liver cirrhosis, skin pigmentation, pancreatic insufficiency, cardiac dysfunction and hypothyroidism[88]. Iron chelation forms the mainstay of therapy in transfusion related siderosis in various hemolytic anemias in children. In a few studies, iron chelators have been implicated in treatment of HH also. Deferoxamine is parenteral iron chelator, given either as subcutaneous or intravenous infusion (20-50 mg/kg per day) over 8-24 h. Adverse effects seen are local reaction in injection site, hearing abnormalities, bone abnormalities etc. Deferasirox is an oral chelator with a similar efficacy as deferoxamine in removing hepatic iron but prone for its gastrointestinal side effects. Deferiprone, also an oral chelator is prone for gastrointestinal side effects and agranulocytosis and is highly effective in removing cardiac iron compared to other chelators (Table 4)[89]. Phatak et al[90] from Italy studied multiple doses of deferoxamine in HH, showed 10 mg/kg is the dose with optimal response and lower side effects. Nagler et al[91] analyzed 2 patients treated for 6 mo and 10 mo respectively who showed significant reduction in serum ferritin in the follow up. EASL and AASLD guidelines on HH recommend phlebotomy as the treatment of choice in HH[92,93]. Chelation may be considered in HH when phlebotomy is not tolerated due to severe congestive cardiac failure, anemia and in case of difficult venous access.
| Properties | Deferoxamine (DFO) | Deferasirox (DFX) | Deferiprone (DFP) |
| Chelator: Iron ratio | 1:1 | 2:1 | 3:1 |
| Plasma t1/2 | 30 min | 12-16 h | 2-3 h |
| Usual dose | 20-50 mg/kg per day over 8-24 h | 20-40 mg/kg per day once daily | 75-100 mg/kg per day in 3 divided doses |
| Route of administration | Subcutaneous, intravenous | Oral | Oral |
| Clearance | Renal, hepatic | Hepatic | Renal |
| Efficacy in removing liver iron stores | Good | Good | Moderate |
| Efficacy in removing cardiac iron | Moderate | Moderate | Good |
| Advantages | Long safety data available, strongest chelator on molar basis | Oral once daily dose is sufficient | Oral, effective in removing cardiac iron |
| Adverse effects | Local reactions | Gastric intolerance | Nausea |
| Sensorineural hearing loss | Rash | Vomiting | |
| Bone abnormalities | Diarrhea | Diarrhea | |
| Retinopathy | Elevation in creatinine | Arthralgia | |
| Pulmonary disease | Elevation in transaminases | Elevated liver enzymes | |
| Allergic reaction | Peptic ulcer | Agranulocytosis | |
| Bacterial infections (e.g., Listeria, Klebsiella) | Renal dysfunction | ||
| Hepatic dysfunction |
In children, secondary hemochromatosis is more common than HH and is usually caused by transfusion related iron overload seen in chronic hemolytic anemia especially beta thalassemia[94]. Each milliliter of packed RBC adds 1mg of iron to the body stores. Iron is usually bound to transferrin in plasma. However when the iron load increases, transferrin sites saturate and excess iron spills as labile plasma iron causing free radical injury to heart, liver and endocrine organs[95]. Multiple transfusion causes liver injury by various mechanisms such as siderosis causing hepatitis eventually progressing to fibrosis and cirrhosis. Hepatic foci of hemopoiesis and transfusion related hepatitis B and C infection are also seen[96].
Iron overload related liver injury can be assessed by various modalities. Serum ferritin is easily available and an inexpensive method to assess iron overload but its utility is limited in the presence of infection and inflammation. Liver iron concentration > 15 mg/g dry weight of liver is associated with significant mortality and morbidity[97]. The superconducting quantum interface device (SQUID) measures liver iron stores non-invasively but the SQUID scanners are not available in many centers worldwide[98]. Magnetic resonance imaging estimates liver iron by R2 and R2* techniques and it correlates well with liver iron concentration attained from biopsy. Magnetic resonance imaging (MRI) has now become the primary monitoring tool for both liver and cardiac iron[99].
Liver injury due to iron overload was common in children in pre-chelation era. Liver biopsies obtained in 80 children with beta thalassemia during splenectomy showed cirrhosis in 40% of children > 11 years with risk of cirrhosis increasing with age. 60% of the children showed hypoalbuminemia and 70% showed elevated transaminases[96]. Iron-chelators are well established treatment modality to prevent iron overload related liver injury. In a retrospective study by Maira et al[100] deferasirox for a duration of 4 ± 1.5 years showed significant improvement in liver stiffness mea
Copper chelation by D-penicillamine and trientine forms the mainstay of treatment in childhood WD. Appropriate dosing, compliance to medications and scheduled monitoring with liver function tests, 24-h urine copper and non- ceruloplasmin copper are required for better control of the disease. D-penicillamine is a promising treatment for Indian childhood cirrhosis especially in early stages. The role in other non-Wilsonian copper diseases is doubtful. The use of iron chelator in Gestational alloimmune liver disease is waning due to its poor efficacy. Iron chelator may be considered as an alternative therapy in hereditary hemochromatosis when the primary treatment fails or not feasible but in case of secondary hemochromatosis chelation forms the main treatment.
Provenance and peer review: Invited article; Externally peer reviewed.
Corresponding Author's Membership in Professional Societies: Indian Society of Gastroenterology.
Specialty type: Gastroenterology and hepatology
Country/Territory of origin: India
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Kanda T, Moschovi MA S-Editor: Ma YJ L-Editor: Filipodia P-Editor: Zhang YL
| 1. | Walshe JM. Penicillamine, a new oral therapy for Wilson's disease. Am J Med. 1956;21:487-495. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 523] [Cited by in RCA: 447] [Article Influence: 6.5] [Reference Citation Analysis (0)] |
| 2. | Mettananda S. Management of thalassaemia. Sri Lanka J Child Heal. 2018;47:159-165. [RCA] [DOI] [Full Text] [Cited by in Crossref: 9] [Cited by in RCA: 9] [Article Influence: 1.3] [Reference Citation Analysis (0)] |
| 3. | Aaseth J, Skaug MA, Cao Y, Andersen O. Chelation in metal intoxication--Principles and paradigms. J Trace Elem Med Biol. 2015;31:260-266. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 114] [Cited by in RCA: 106] [Article Influence: 10.6] [Reference Citation Analysis (0)] |
| 4. | Al-Karadaghi S, Franco R, Hansson M, Shelnutt JA, Isaya G, Ferreira GC. Chelatases: distort to select? Trends Biochem Sci.. 2006;31(3):135-42. [RCA] [DOI] [Full Text] [Cited by in RCA: 1] [Reference Citation Analysis (0)] |
| 5. | Flora SJ, Pachauri V. Chelation in metal intoxication. Int J Environ Res Public Health. 2010;7:2745-2788. [RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)] [Cited by in Crossref: 648] [Cited by in RCA: 561] [Article Influence: 37.4] [Reference Citation Analysis (0)] |
| 6. | de Bie P, Muller P, Wijmenga C, Klomp LW. Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes. J Med Genet. 2007;44:673-688. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 266] [Cited by in RCA: 259] [Article Influence: 14.4] [Reference Citation Analysis (0)] |
| 7. | Lalioti V, Sandoval I, Cassio D, Duclos-Vallée JC. Molecular pathology of Wilson's disease: a brief. J Hepatol. 2010;53:1151-1153. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 26] [Cited by in RCA: 24] [Article Influence: 1.6] [Reference Citation Analysis (0)] |
| 8. | Peisach J, Blumberg WE. A mechanism for the action of penicillamine in the treatment of Wilson's disease. Mol Pharmacol. 1969;5:200-209. [PubMed] |
| 9. | Kukovetz WR, Beubler E, Kreuzig F, Moritz AJ, Nirnberger G, Werner-Breitenecker L. Bioavailability and pharmacokinetics of D-penicillamine. J Rheumatol. 1983;10:90-94. [PubMed] |
| 10. | Netter P, Bannwarth B, Péré P, Nicolas A. Clinical pharmacokinetics of D-penicillamine. Clin Pharmacokinet. 1987;13:317-333. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 50] [Cited by in RCA: 48] [Article Influence: 1.3] [Reference Citation Analysis (0)] |
| 11. | Langlois DK, Lehner AF, Buchweitz JP, Ross DE, Johnson MB, Kruger JM, Bailie MB, Hauptman JG, Schall WD. Pharmacokinetics and relative bioavailability of D-penicillamine in fasted and nonfasted dogs. J Vet Intern Med. 2013;27:1071-1076. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 9] [Cited by in RCA: 8] [Article Influence: 0.7] [Reference Citation Analysis (0)] |
| 12. | Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47:2089-2111. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 884] [Cited by in RCA: 812] [Article Influence: 47.8] [Reference Citation Analysis (0)] |
| 13. | Litwin T, Dzieżyc K, Karliński M, Chabik G, Czepiel W, Członkowska A. Early neurological worsening in patients with Wilson's disease. J Neurol Sci. 2015;355:162-167. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 89] [Cited by in RCA: 97] [Article Influence: 9.7] [Reference Citation Analysis (0)] |
| 14. | Lu J. Triethylenetetramine pharmacology and its clinical applications. Mol Cancer Ther. 2010;9:2458-2467. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 40] [Cited by in RCA: 44] [Article Influence: 2.9] [Reference Citation Analysis (0)] |
| 15. | European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson's disease. J Hepatol. 2012;56:671-685. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 971] [Cited by in RCA: 781] [Article Influence: 60.1] [Reference Citation Analysis (1)] |
| 16. | Smirnova J, Kabin E, Järving I, Bragina O, Tõugu V, Plitz T, Palumaa P. Copper(I)-binding properties of de-coppering drugs for the treatment of Wilson disease. α-Lipoic acid as a potential anti-copper agent. Sci Rep. 2018;8:1463. [RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)] [Cited by in Crossref: 36] [Cited by in RCA: 56] [Article Influence: 8.0] [Reference Citation Analysis (0)] |
| 17. | Lau JY, Lai CL, Wu PC, Pan HY, Lin HJ, Todd D. Wilson's disease: 35 years' experience. Q J Med. 1990;75:597-605. [PubMed] |
| 18. | Svetel M, Pekmezović T, Petrović I, Tomić A, Kresojević N, Jesić R, Kazić S, Raicević R, Stefanović D, Delibasić N, Zivanović D, Dordević M, Kostić VS. Long-term outcome in Serbian patients with Wilson disease. Eur J Neurol. 2009;16:852-857. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 34] [Cited by in RCA: 34] [Article Influence: 2.1] [Reference Citation Analysis (0)] |
| 19. | Bruha R, Marecek Z, Pospisilova L, Nevsimalova S, Vitek L, Martasek P, Nevoral J, Petrtyl J, Urbanek P, Jiraskova A, Ferenci P. Long-term follow-up of Wilson disease: natural history, treatment, mutations analysis and phenotypic correlation. Liver Int. 2011;31:83-91. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 112] [Cited by in RCA: 95] [Article Influence: 6.8] [Reference Citation Analysis (0)] |
| 20. | Weiss KH, Thurik F, Gotthardt DN, Schäfer M, Teufel U, Wiegand F, Merle U, Ferenci-Foerster D, Maieron A, Stauber R, Zoller H, Schmidt HH, Reuner U, Hefter H, Trocello JM, Houwen RH, Ferenci P, Stremmel W; EUROWILSON Consortium. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013;11:1028-35.e1. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 186] [Cited by in RCA: 137] [Article Influence: 11.4] [Reference Citation Analysis (0)] |
| 21. | Walshe JM. Treatment of Wilson's disease with trientine (triethylene tetramine) dihydrochloride. Lancet. 1982;1:643-647. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 272] [Cited by in RCA: 234] [Article Influence: 5.4] [Reference Citation Analysis (0)] |
| 22. | Scheinberg IH, Jaffe ME, Sternlieb I. The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson's disease. N Engl J Med. 1987;317:209-213. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 153] [Cited by in RCA: 122] [Article Influence: 3.2] [Reference Citation Analysis (0)] |
| 23. | Hölscher S, Leinweber B, Hefter H, Reuner U, Günther P, Weiss KH, Oertel WH, Möller JC. Evaluation of the symptomatic treatment of residual neurological symptoms in Wilson disease. Eur Neurol. 2010;64:83-87. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 30] [Cited by in RCA: 29] [Article Influence: 1.9] [Reference Citation Analysis (0)] |
| 24. | Medici V, Trevisan CP, D'Incà R, Barollo M, Zancan L, Fagiuoli S, Martines D, Irato P, Sturniolo GC. Diagnosis and management of Wilson's disease: results of a single center experience. J Clin Gastroenterol. 2006;40:936-941. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 119] [Cited by in RCA: 101] [Article Influence: 5.3] [Reference Citation Analysis (0)] |
| 25. | Czlonkowska A, Gajda J, Rodo M. Effects of long-term treatment in Wilson's disease with D-penicillamine and zinc sulphate. J Neurol. 1996;243:269-273. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 133] [Cited by in RCA: 108] [Article Influence: 3.7] [Reference Citation Analysis (0)] |
| 26. | LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012 . [PubMed] |
| 27. | Hall CL, Jawad S, Harrison PR, MacKenzie JC, Bacon PA, Klouda PT, MacIver AG. Natural course of penicillamine nephropathy: a long term study of 33 patients. Br Med J (Clin Res Ed). 1988;296:1083-1086. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 55] [Cited by in RCA: 48] [Article Influence: 1.3] [Reference Citation Analysis (0)] |
| 28. | Bienaimé F, Clerbaux G, Plaisier E, Mougenot B, Ronco P, Rougier JP. D-Penicillamine-induced ANCA-associated crescentic glomerulonephritis in Wilson disease. Am J Kidney Dis. 2007;50:821-825. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 29] [Cited by in RCA: 33] [Article Influence: 1.8] [Reference Citation Analysis (0)] |
| 29. | Derk CT, Jimenez SA. Goodpasture-like syndrome induced by D-penicillamine in a patient with systemic sclerosis: report and review of the literature. J Rheumatol. 2003;30:1616-1620. [PubMed] |
| 30. | Billingsley LM, Stevens MB. The relationship between D-penicillamine--induced proteinuria and prior gold nephropathy. Johns Hopkins Med J. 1981;148:64-67. [PubMed] |
| 31. | Steen VD, Blair S, Medsger TA Jr. The toxicity of D-penicillamine in systemic sclerosis. Ann Intern Med. 1986;104:699-705. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 67] [Cited by in RCA: 57] [Article Influence: 1.5] [Reference Citation Analysis (0)] |
| 32. | Toxicity of longterm low dose D-penicillamine therapy in rheumatoid arthritis. Cooperative Systematic Studies of Rheumatic Disease Group. J Rheumatol. 1987;14:67-73. [PubMed] |
| 33. | Kay AG. Myelotoxicity of D-penicillamine. Ann Rheum Dis. 1979;38:232-236. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 33] [Cited by in RCA: 31] [Article Influence: 0.7] [Reference Citation Analysis (0)] |
| 34. | Jaffe IA. Adverse effects profile of sulfhydryl compounds in man. Am J Med. 1986;80:471-476. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 115] [Cited by in RCA: 99] [Article Influence: 2.5] [Reference Citation Analysis (0)] |
| 35. | Gupta P, Choksi M, Goel A, Zachariah U, Sajith KG, Ramachandran J, Chandy G, Kurian G, Rebekah G, Eapen CE. Maintenance zinc therapy after initial penicillamine chelation to treat symptomatic hepatic Wilson's disease in resource constrained setting. Indian J Gastroenterol. 2018;37:31-38. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 10] [Cited by in RCA: 15] [Article Influence: 2.1] [Reference Citation Analysis (0)] |
| 36. | Pitman SK, Huynh T, Bjarnason TA, An J, Malkhasyan KA. A case report and focused literature review of d-penicillamine and severe neutropenia: A serious toxicity from a seldom-used drug. Clin Case Rep. 2019;7:990-994. [RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)] [Cited by in Crossref: 6] [Cited by in RCA: 9] [Article Influence: 1.5] [Reference Citation Analysis (0)] |
| 37. | Petrides PE, Gerhartz HH. D-penicillamine-induced agranulocytosis: hematological remission upon treatment with recombinant GM-CSF. Z Rheumatol. 1991;50:328-329. [PubMed] |
| 38. | Ishak R, Abbas O. Penicillamine revisited: historic overview and review of the clinical uses and cutaneous adverse effects. Am J Clin Dermatol. 2013;14:223-233. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 33] [Cited by in RCA: 38] [Article Influence: 3.2] [Reference Citation Analysis (0)] |
| 39. | Nishida H, Sahashi K. [Penicillamine-induced myasthenia gravis]. Ryoikibetsu Shokogun Shirizu. 2001;351-353. [PubMed] |
| 40. | Lee Y, Lee ST, Cho H. D-penicillamine-induced ANA (+) ANCA (+) vasculitis in pediatric patients with Wilson's disease. Clin Nephrol. 2016;85:296-300. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 11] [Cited by in RCA: 11] [Article Influence: 1.2] [Reference Citation Analysis (0)] |
| 41. | Deutscher J, Kiess W, Scheerschmidt G, Willgerodt H. Potential hepatotoxicity of penicillamine treatment in three patients with Wilson's disease. J Pediatr Gastroenterol Nutr. 1999;29:628. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 16] [Cited by in RCA: 13] [Article Influence: 0.5] [Reference Citation Analysis (0)] |
| 42. | Kalita J, Kumar V, Ranjan A, Misra UK. Role of Oxidative Stress in the Worsening of Neurologic Wilson Disease Following Chelating Therapy. Neuromolecular Med. 2015;17:364-372. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 17] [Cited by in RCA: 21] [Article Influence: 2.1] [Reference Citation Analysis (0)] |
| 43. | Cramer JA, Roy A, Burrell A, Fairchild CJ, Fuldeore MJ, Ollendorf DA, Wong PK. Medication compliance and persistence: terminology and definitions. Value Health. 2008;11:44-47. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 1363] [Cited by in RCA: 1561] [Article Influence: 91.8] [Reference Citation Analysis (0)] |
| 44. | Masełbas W, Członkowska A, Litwin T, Niewada M. Persistence with treatment for Wilson disease: a retrospective study. BMC Neurol. 2019;19:278. [RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)] [Cited by in Crossref: 23] [Cited by in RCA: 36] [Article Influence: 6.0] [Reference Citation Analysis (0)] |
| 45. | Lössner A, Lössner J, Bachmann H, Zotter J. The Kayser-Fleischer ring during long-term treatment in Wilson's disease (hepatolenticular degeneration). A follow-up study. Graefes Arch Clin Exp Ophthalmol. 1986;224:152-155. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 26] [Cited by in RCA: 19] [Article Influence: 0.5] [Reference Citation Analysis (0)] |
| 46. | Fenu M, Liggi M, Demelia E, Sorbello O, Civolani A, Demelia L. Kayser-Fleischer ring in Wilson's disease: a cohort study. Eur J Intern Med. 2012;23:e150-e156. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 21] [Cited by in RCA: 22] [Article Influence: 1.7] [Reference Citation Analysis (0)] |
| 47. | Esmaeli B, Burnstine MA, Martonyi CL, Sugar A, Johnson V, Brewer GJ. Regression of Kayser-Fleischer rings during oral zinc therapy: correlation with systemic manifestations of Wilson's disease. Cornea. 1996;15:582-588. [PubMed] |
| 48. | Marcellini M, Di Ciommo V, Callea F, Devito R, Comparcola D, Sartorelli MR, Carelli G, Nobili V. Treatment of Wilson's disease with zinc from the time of diagnosis in pediatric patients: a single-hospital, 10-year follow-up study. J Lab Clin Med. 2005;145:139-143. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 85] [Cited by in RCA: 74] [Article Influence: 3.7] [Reference Citation Analysis (0)] |
| 49. | Suvarna JC. Kayser-Fleischer ring. J Postgrad Med. 2008;54:238-240. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 35] [Cited by in RCA: 31] [Article Influence: 1.8] [Reference Citation Analysis (0)] |
| 50. | Das MC, Sen Sarma M, Srivastava A, Yachha SK, Poddar U. Effect of chelation therapy in pediatric Wilson's disease: Liver and endoscopic outcome. J Hepatobiliary Pancreat Sci. 2021;28:336-345. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 5] [Cited by in RCA: 10] [Article Influence: 2.0] [Reference Citation Analysis (0)] |
| 51. | Aggarwal A, Aggarwal N, Nagral A, Jankharia G, Bhatt M. A novel Global Assessment Scale for Wilson's Disease (GAS for WD). Mov Disord. 2009;24:509-518. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 70] [Cited by in RCA: 84] [Article Influence: 5.3] [Reference Citation Analysis (0)] |
| 52. | Prashanth LK, Taly AB, Sinha S, Ravishankar S, Arunodaya GR, Vasudev MK, Swamy HS. Prognostic factors in patients presenting with severe neurological forms of Wilson's disease. QJM. 2005;98:557-563. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 43] [Cited by in RCA: 42] [Article Influence: 2.1] [Reference Citation Analysis (0)] |
| 53. | Walshe JM. The pattern of urinary copper excretion and its response to treatment in patients with Wilson's disease. QJM. 2011;104:775-778. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 29] [Cited by in RCA: 31] [Article Influence: 2.2] [Reference Citation Analysis (0)] |
| 54. | Twomey PJ, Viljoen A, Reynolds TM, Wierzbicki AS. Non-ceruloplasmin-bound copper in routine clinical practice in different laboratories. J Trace Elem Med Biol. 2008;22:50-53. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 26] [Cited by in RCA: 30] [Article Influence: 1.8] [Reference Citation Analysis (0)] |
| 55. | Duncan A, Yacoubian C, Beetham R, Catchpole A, Bullock D. The role of calculated non-caeruloplasmin-bound copper in Wilson's disease. Ann Clin Biochem. 2017;54:649-654. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 12] [Cited by in RCA: 12] [Article Influence: 1.3] [Reference Citation Analysis (0)] |
| 56. | Schmitt F, Podevin G, Poupon J, Roux J, Legras P, Trocello JM, Woimant F, Laprévote O, Nguyen TH, El Balkhi S. Evolution of exchangeable copper and relative exchangeable copper through the course of Wilson's disease in the Long Evans Cinnamon rat. PLoS One. 2013;8:e82323. [RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)] [Cited by in Crossref: 23] [Cited by in RCA: 18] [Article Influence: 1.5] [Reference Citation Analysis (0)] |
| 57. | UEG Week 2020 Poster Presentations. United European Gastroenterol J. 2020;8:144-887. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 6] [Cited by in RCA: 1] [Article Influence: 0.2] [Reference Citation Analysis (0)] |
| 58. | Cousins RJ. Absorption, transport, and hepatic metabolism of copper and zinc: special reference to metallothionein and ceruloplasmin. Physiol Rev. 1985;65:238-309. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 892] [Cited by in RCA: 812] [Article Influence: 20.3] [Reference Citation Analysis (0)] |
| 59. | Santos Silva EE, Sarles J, Buts JP, Sokal EM. Successful medical treatment of severely decompensated Wilson disease. J Pediatr. 1996;128:285-287. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 65] [Cited by in RCA: 45] [Article Influence: 1.6] [Reference Citation Analysis (0)] |
| 60. | Dhawan A, Taylor RM, Cheeseman P, De Silva P, Katsiyiannakis L, Mieli-Vergani G. Wilson's disease in children: 37-year experience and revised King's score for liver transplantation. Liver Transpl. 2005;11:441-448. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 296] [Cited by in RCA: 253] [Article Influence: 12.7] [Reference Citation Analysis (0)] |
| 61. | Askari FK, Greenson J, Dick RD, Johnson VD, Brewer GJ. Treatment of Wilson's disease with zinc. XVIII. Initial treatment of the hepatic decompensation presentation with trientine and zinc. J Lab Clin Med. 2003;142:385-390. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 89] [Cited by in RCA: 63] [Article Influence: 3.0] [Reference Citation Analysis (0)] |
| 62. | Chen JC, Chuang CH, Wang JD, Wang CW. Combination Therapy Using Chelating Agent and Zinc for Wilson's Disease. J Med Biol Eng. 2015;35:697-708. [RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)] [Cited by in Crossref: 19] [Cited by in RCA: 12] [Article Influence: 1.2] [Reference Citation Analysis (0)] |
| 63. | Weiss KH, Gotthardt DN, Klemm D, Merle U, Ferenci-Foerster D, Schaefer M, Ferenci P, Stremmel W. Zinc monotherapy is not as effective as chelating agents in treatment of Wilson disease. Gastroenterology. 2011;140:1189-1198.e1. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 172] [Cited by in RCA: 124] [Article Influence: 8.9] [Reference Citation Analysis (0)] |
| 64. | Fieten H, Dirksen K, van den Ingh TS, Winter EA, Watson AL, Leegwater PA, Rothuizen J. D-penicillamine treatment of copper-associated hepatitis in Labrador retrievers. Vet J. 2013;196:522-527. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 24] [Cited by in RCA: 28] [Article Influence: 2.3] [Reference Citation Analysis (0)] |
| 65. | Brewer GJ, Hedera P, Kluin KJ, Carlson M, Askari F, Dick RB, Sitterly J, Fink JK. Treatment of Wilson disease with ammonium tetrathiomolybdate: III. Initial therapy in a total of 55 neurologically affected patients and follow-up with zinc therapy. Arch Neurol. 2003;60:379-385. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 160] [Cited by in RCA: 123] [Article Influence: 5.6] [Reference Citation Analysis (0)] |
| 66. | Brewer GJ, Askari F, Lorincz MT, Carlson M, Schilsky M, Kluin KJ, Hedera P, Moretti P, Fink JK, Tankanow R, Dick RB, Sitterly J. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol. 2006;63:521-527. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 215] [Cited by in RCA: 191] [Article Influence: 10.1] [Reference Citation Analysis (0)] |
| 67. | Medici V, Trevisan CP, Bigotto MA, D'Incà R, Martines D, Dal Pont E, Sturniolo GC. Adverse reaction after tetrathiomolybdate treatment for Wilson's disease: a case report. Mov Disord. 2006;21:2030-2032. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 12] [Cited by in RCA: 11] [Article Influence: 0.6] [Reference Citation Analysis (0)] |
| 68. | Karunajeewa H, Wall A, Metz J, Grigg A. Cytopenias secondary to copper depletion complicating ammonium tetrathiomolybdate therapy for Wilson's disease. Aust N Z J Med. 1998;28:215-216. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 20] [Cited by in RCA: 20] [Article Influence: 0.7] [Reference Citation Analysis (0)] |
| 69. | Weiss KH, Askari FK, Czlonkowska A, Ferenci P, Bronstein JM, Bega D, Ala A, Nicholl D, Flint S, Olsson L, Plitz T, Bjartmar C, Schilsky ML. Bis-choline tetrathiomolybdate in patients with Wilson's disease: an open-label, multicentre, phase 2 study. Lancet Gastroenterol Hepatol. 2017;2:869-876. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 124] [Cited by in RCA: 92] [Article Influence: 11.5] [Reference Citation Analysis (0)] |
| 70. | Pandit A, Bhave S. Present interpretation of the role of copper in Indian childhood cirrhosis. Am J Clin Nutr. 1996;63:830S-835S. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 60] [Cited by in RCA: 47] [Article Influence: 1.6] [Reference Citation Analysis (0)] |
| 71. | Chawla V, Chandra RK, Verma IC, Ghai OP. An epidemiologic approach to Indian childhood cirrhosis. Indian Pediatr. 1973;10:73-79. [PubMed] |
| 72. | Nayak NC, Visalakshi S, Singh M, Chawla V, Chandra RK, Ramalingaswami V. Indian childhood cirrhosis--a re-evaluation of its pathomorphologic features and their significance in the light of clinical data and natural history of the disease. Indian J Med Res. 1972;60:246-259. [PubMed] |
| 73. | Bavdekar AR, Bhave SA, Pradhan AM, Pandit AN, Tanner MS. Long term survival in Indian childhood cirrhosis treated with D-penicillamine. Arch Dis Child. 1996;74:32-35. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 21] [Cited by in RCA: 15] [Article Influence: 0.5] [Reference Citation Analysis (0)] |
| 74. | Tanner MS, Bhave SA, Pradhan AM, Pandit AN. Clinical trials of penicillamine in Indian childhood cirrhosis. Arch Dis Child. 1987;62:1118-1124. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 31] [Cited by in RCA: 23] [Article Influence: 0.6] [Reference Citation Analysis (0)] |
| 75. | Tomar BS, Saxena S, Prakash P, Tomar S, Verma C. D-penicillamine in the treatment of Indian childhood cirrhosis--a preliminary report. Indian J Pediatr. 1983;50:613-618. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 7] [Cited by in RCA: 7] [Article Influence: 0.2] [Reference Citation Analysis (0)] |
| 76. | Baker A, Gormally S, Saxena R, Baldwin D, Drumm B, Bonham J, Portmann B, Mowat AP. Copper-associated liver disease in childhood. J Hepatol. 1995;23:538-543. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 38] [Cited by in RCA: 24] [Article Influence: 0.8] [Reference Citation Analysis (0)] |
| 77. | Horslen SP, Tanner MS, Lyon TD, Fell GS, Lowry MF. Copper associated childhood cirrhosis. Gut. 1994;35:1497-1500. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 28] [Cited by in RCA: 26] [Article Influence: 0.8] [Reference Citation Analysis (0)] |
| 78. | Maggiore G, De Giacomo C, Sessa F, Burgio GR. Idiopathic hepatic copper toxicosis in a child. J Pediatr Gastroenterol Nutr. 1987;6:980-983. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 25] [Cited by in RCA: 21] [Article Influence: 0.6] [Reference Citation Analysis (0)] |
| 79. | Muller T, Feichtinger H, Berger H, Muller W. Endemic Tyrolean infantile cirrhosis: an ecogenetic disorder. Lancet. 1996;347:877-880. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 144] [Cited by in RCA: 95] [Article Influence: 3.3] [Reference Citation Analysis (0)] |
| 80. | Kelly AL, Lunt PW, Rodrigues F, Berry PJ, Flynn DM, McKiernan PJ, Kelly DA, Mieli-Vergani G, Cox TM. Classification and genetic features of neonatal haemochromatosis: a study of 27 affected pedigrees and molecular analysis of genes implicated in iron metabolism. J Med Genet. 2001;38:599-610. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 83] [Cited by in RCA: 60] [Article Influence: 2.5] [Reference Citation Analysis (0)] |
| 81. | Feldman AG, Whitington PF. Neonatal hemochromatosis. J Clin Exp Hepatol. 2013;3:313-320. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 71] [Cited by in RCA: 77] [Article Influence: 6.4] [Reference Citation Analysis (0)] |
| 82. | Lopriore E, Mearin ML, Oepkes D, Devlieger R, Whitington PF. Neonatal hemochromatosis: management, outcome, and prevention. Prenat Diagn. 2013;33:1221-1225. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 49] [Cited by in RCA: 41] [Article Influence: 3.4] [Reference Citation Analysis (0)] |
| 83. | Flynn DM, Mohan N, McKiernan P, Beath S, Buckels J, Mayer D, Kelly DA. Progress in treatment and outcome for children with neonatal haemochromatosis. Arch Dis Child Fetal Neonatal Ed. 2003;88:F124-F127. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 81] [Cited by in RCA: 65] [Article Influence: 3.0] [Reference Citation Analysis (0)] |
| 84. | Rodrigues F, Kallas M, Nash R, Cheeseman P, D'Antiga L, Rela M, Heaton ND, Mieli-Vergani G. Neonatal hemochromatosis--medical treatment vs. transplantation: the king's experience. Liver Transpl. 2005;11:1417-1424. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 74] [Cited by in RCA: 60] [Article Influence: 3.0] [Reference Citation Analysis (0)] |
| 85. | Sigurdsson L, Reyes J, Kocoshis SA, Hansen TW, Rosh J, Knisely AS. Neonatal hemochromatosis: outcomes of pharmacologic and surgical therapies. J Pediatr Gastroenterol Nutr. 1998;26:85-89. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 87] [Cited by in RCA: 68] [Article Influence: 2.5] [Reference Citation Analysis (0)] |
| 86. | Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393-408, 408.e1. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 390] [Cited by in RCA: 388] [Article Influence: 25.9] [Reference Citation Analysis (0)] |
| 87. | Higuchi T, Moriyama M, Fukushima A, Matsumura H, Matsuoka S, Kanda T, Sugitani M, Tsunemi A, Ueno T, Fukuda N. Association of mRNA expression of iron metabolism-associated genes and progression of non-alcoholic steatohepatitis in rats. Oncotarget. 2018;9:26183-26194. [RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)] [Cited by in Crossref: 11] [Cited by in RCA: 9] [Article Influence: 1.3] [Reference Citation Analysis (0)] |
| 88. | Salgia RJ, Brown K. Diagnosis and management of hereditary hemochromatosis. Clin Liver Dis. 2015;19:187-198. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 42] [Cited by in RCA: 37] [Article Influence: 3.7] [Reference Citation Analysis (0)] |
| 89. | Mobarra N, Shanaki M, Ehteram H, Nasiri H, Sahmani M, Saeidi M, Goudarzi M, Pourkarim H, Azad M. A Review on Iron Chelators in Treatment of Iron Overload Syndromes. Int J Hematol Oncol Stem Cell Res. 2016;10:239-247. [PubMed] |
| 90. | Phatak P, Brissot P, Wurster M, Adams PC, Bonkovsky HL, Gross J, Malfertheiner P, McLaren GD, Niederau C, Piperno A, Powell LW, Russo MW, Stoelzel U, Stremmel W, Griffel L, Lynch N, Zhang Y, Pietrangelo A. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology. 2010;52:1671-1779. [RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)] [Cited by in Crossref: 83] [Cited by in RCA: 78] [Article Influence: 5.2] [Reference Citation Analysis (0)] |
| 91. | Nagler M, Gregor M, Wuillemin WA. Iron chelation with deferasirox in two patients with HFE hemochromatosis and chronic anemia. Acta Haematol. 2011;126:119-121. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 5] [Cited by in RCA: 6] [Article Influence: 0.4] [Reference Citation Analysis (0)] |
| 92. | Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328-343. [RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)] [Cited by in Crossref: 421] [Cited by in RCA: 422] [Article Influence: 30.1] [Reference Citation Analysis (0)] |
| 93. | European Association for the Study of the Liver. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53:3-22. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 416] [Cited by in RCA: 352] [Article Influence: 23.5] [Reference Citation Analysis (0)] |
| 94. | Ware HM, Kwiatkowski JL. Evaluation and treatment of transfusional iron overload in children. Pediatr Clin North Am. 2013;60:1393-1406. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 30] [Cited by in RCA: 35] [Article Influence: 2.9] [Reference Citation Analysis (0)] |
| 95. | Piga A, Longo F, Duca L, Roggero S, Vinciguerra T, Calabrese R, Hershko C, Cappellini MD. High nontransferrin bound iron levels and heart disease in thalassemia major. Am J Hematol. 2009;84:29-33. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 104] [Cited by in RCA: 115] [Article Influence: 7.2] [Reference Citation Analysis (0)] |
| 96. | Jean G, Terzoli S, Mauri R, Borghetti L, Di Palma A, Piga A, Magliano M, Melevendi M, Cattaneo M. Cirrhosis associated with multiple transfusions in thalassaemia. Arch Dis Child. 1984;59:67-70. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 42] [Cited by in RCA: 35] [Article Influence: 0.9] [Reference Citation Analysis (0)] |
| 97. | Olivieri NF, Nathan DG, MacMillan JH, Wayne AS, Liu PP, McGee A, Martin M, Koren G, Cohen AR. Survival in medically treated patients with homozygous beta-thalassemia. N Engl J Med. 1994;331:574-578. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 624] [Cited by in RCA: 629] [Article Influence: 20.3] [Reference Citation Analysis (0)] |
| 98. | Brittenham GM, Farrell DE, Harris JW, Feldman ES, Danish EH, Muir WA, Tripp JH, Bellon EM. Magnetic-susceptibility measurement of human iron stores. N Engl J Med. 1982;307:1671-1675. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 280] [Cited by in RCA: 253] [Article Influence: 5.9] [Reference Citation Analysis (0)] |
| 99. | Wood JC, Enriquez C, Ghugre N, Tyzka JM, Carson S, Nelson MD, Coates TD. MRI R2 and R2* mapping accurately estimates hepatic iron concentration in transfusion-dependent thalassemia and sickle cell disease patients. Blood. 2005;106:1460-1465. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 794] [Cited by in RCA: 831] [Article Influence: 41.6] [Reference Citation Analysis (0)] |
| 100. | Maira D, Cassinerio E, Marcon A, Mancarella M, Fraquelli M, Pedrotti P, Cappellini MD. Progression of liver fibrosis can be controlled by adequate chelation in transfusion-dependent thalassemia (TDT). Ann Hematol. 2017;96:1931-1936. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 16] [Cited by in RCA: 12] [Article Influence: 1.5] [Reference Citation Analysis (0)] |
| 101. | Piga A, Longo F, Musallam KM, Cappellini MD, Forni GL, Quarta G, Chiavilli F, Commendatore F, Mulas S, Caruso V, Galanello R. Assessment and management of iron overload in β-thalassaemia major patients during the 21st century: a real-life experience from the Italian WEBTHAL project. Br J Haematol. 2013;161:872-883. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 23] [Cited by in RCA: 31] [Article Influence: 2.6] [Reference Citation Analysis (0)] |
| 102. | Angelucci E, Barosi G, Camaschella C, Cappellini MD, Cazzola M, Galanello R, Marchetti M, Piga A, Tura S. Italian Society of Hematology practice guidelines for the management of iron overload in thalassemia major and related disorders. Haematologica. 2008;93:741-752. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 137] [Cited by in RCA: 143] [Article Influence: 8.4] [Reference Citation Analysis (0)] |
| 103. | Kamble RT, Selby GB, Mims M, Kharfan-Dabaja MA, Ozer H, George JN. Iron overload manifesting as apparent exacerbation of hepatic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2006;12:506-510. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 59] [Cited by in RCA: 59] [Article Influence: 3.1] [Reference Citation Analysis (0)] |
| 104. | Majhail NS, Lazarus HM, Burns LJ. Iron overload in hematopoietic cell transplantation. Bone Marrow Transplant. 2008;41:997-1003. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 97] [Cited by in RCA: 103] [Article Influence: 6.1] [Reference Citation Analysis (0)] |
| 105. | Sivgin S, Eser B. The management of iron overload in allogeneic hematopoietic stem cell transplant (alloHSCT) recipients: where do we stand? Ann Hematol. 2013;92:577-586. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 19] [Cited by in RCA: 22] [Article Influence: 1.8] [Reference Citation Analysis (0)] |
| 106. | Wang LC, Wang JD, Tsai CR, Cheng SB, Lin CC. Clinical features and therapeutic response in Taiwanese children with Wilson's disease: 12 years of experience in a single center. Pediatr Neonatol. 2010;51:124-129. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 12] [Cited by in RCA: 12] [Article Influence: 0.8] [Reference Citation Analysis (0)] |
| 107. | Arnon R, Calderon JF, Schilsky M, Emre S, Shneider BL. Wilson disease in children: serum aminotransferases and urinary copper on triethylene tetramine dihydrochloride (trientine) treatment. J Pediatr Gastroenterol Nutr. 2007;44:596-602. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 48] [Cited by in RCA: 38] [Article Influence: 2.1] [Reference Citation Analysis (0)] |
| 108. | Taylor RM, Chen Y, Dhawan A; EuroWilson Consortium. Triethylene tetramine dihydrochloride (trientine) in children with Wilson disease: experience at King's College Hospital and review of the literature. Eur J Pediatr. 2009;168:1061-1068. [RCA] [PubMed] [DOI] [Full Text] [Cited by in Crossref: 41] [Cited by in RCA: 31] [Article Influence: 1.9] [Reference Citation Analysis (0)] |
| 109. | Rodeck B, Kardoff R, Melter M. Treatment of copper associated liver disease in childhood. Eur J Med Res. 1999;4:253-256. [PubMed] |
| 110. | Masera N, Cattoni A, Decimi V, D’Apolito Valeria, Arosio C, Mariani R, Piperno A. Efficacy of deferasirox for the treatment of iron overload in a child affected by Juvenile Hemochromatosis. Case Rep Clin Med. 2013;2:126-128. [RCA] [DOI] [Full Text] [Cited by in Crossref: 1] [Cited by in RCA: 1] [Article Influence: 0.1] [Reference Citation Analysis (0)] |